Deuterium‐reinforced linoleic acid lowers lipid peroxidation and mitigates cognitive impairment in the Q140 knock in mouse model of Huntington’s disease
Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease which has no effective treatment and is characterized by psychiatric disorders, motor alterations, and dementia, with the cognitive deficits representing a devastating aspect of the disorder. Oxidative stress and elevated levels of lipid peroxidation (LPO) products are found in mouse models and patients with HD, suggesting that strategies to reduce LPO may be beneficial in HD. In contrast with traditional antioxidants, substituting hydrogen with deuterium at bis‐allylic sites in polyunsaturated fatty acids (D‐PUFA) decreases the rate‐limiting initiation step of PUFA autoxidation, a strategy that has shown benefits in other neurodegenerative diseases. Here, we investigated the effect of D‐PUFA treatment in a knock‐in mouse model of HD (Q140) which presents motor deficits and neuropathology from a few months of age, and progressive cognitive decline. Q140 knock‐in mice were fed a diet containing either D‐ or H‐PUFAs for 5 months starting at 1 month of age. D‐PUFA treatment significantly decreased F2‐isoprostanes in the striatum by approximately 80% as compared to H‐PUFA treatment and improved performance in novel object recognition tests, without significantly changing motor deficits or huntingtin aggregation. Therefore, D‐PUFA administration represents a promising new strategy to broadly reduce rates of LPO, and may be useful in improving a subset of the core deficits in HD.
Abbreviations
ARA
arachidonic
D‐PUFA
Deuterated polyunsaturated fatty acids
HD
Huntington’s disease
LPO
lipid peroxidation
Introduction
Oxidative damage due to increased oxidative stress may be an important factor in the pathogenesis of Huntington’s disease (HD) and other neurodegenerative diseases including retinal diseases, Parkinson’s disease (PD), and Alzheimer’s disease. High brain levels of polyunsaturated fatty acids (PUFAs) and transition metals, coupled with high oxygen utilization and modest antioxidant defense, create an environment especially vulnerable to oxidative damage . HD is a fatal disease causing progressive degeneration of selected neuronal populations, primarily in the striatum and the cortex. The mutation responsible for HD is an abnormally (above 39) expanded and unstable CAG (encoding glutamine) repeat within the coding region of the gene encoding huntingtin. Mitochondrial dysfunction is an important aspect of HD. Oxidative damage is present in the very early stages of HD, suggesting that it may contribute to HD pathogenesis.
Reactive oxygen species (ROS)‐initiated nonenzymatic lipid peroxidation (LPO) of PUFAs such as linoleic (Lin), linolenic (Lnn), arachidonic (ARA), eicosapentaenoic (EPA), and docosahexaenoic (DHA) acids is the major contributor to oxidative damage, resulting in altered membrane fluidity and changes in membrane‐bound enzymes and receptors. LPO of PUFAs by ROS is initiated by abstraction of bis‐allylic (between double bonds) hydrogens, generating resonance‐stabilized free radicals that subsequently react with molecular oxygen to form lipid radicals (Fig. 1). These newly formed species abstract a hydrogen atom of an adjacent PUFA molecule, thus propagating a chain reaction. Propagation is eventually terminated by a chain‐terminating antioxidant such as vitamin E or by recombination of two radicals. LPO results in the formation of two broad categories of secondary byproducts derived through either endoperoxide or hydroperoxide intermediates. Examples of the former are F2‐isoprostanes (F2‐isoP) derived from ARA, EPA‐derived F3 isoprostanes, and F4‐isoprostanes (neuroprostanes) derived from DHA. The chemical stability of these metabolites has led to their use as reliable biomarkers for monitoring LPO. Decomposition of hydroperoxide intermediates gives rise to a number of reactive aldehydes, such as malondialdehyde (MDA), acrolein, 4‐hydroxy‐2‐nonenal (HNE), 4‐hydroxy‐2‐hexenal (HHE), and many others.
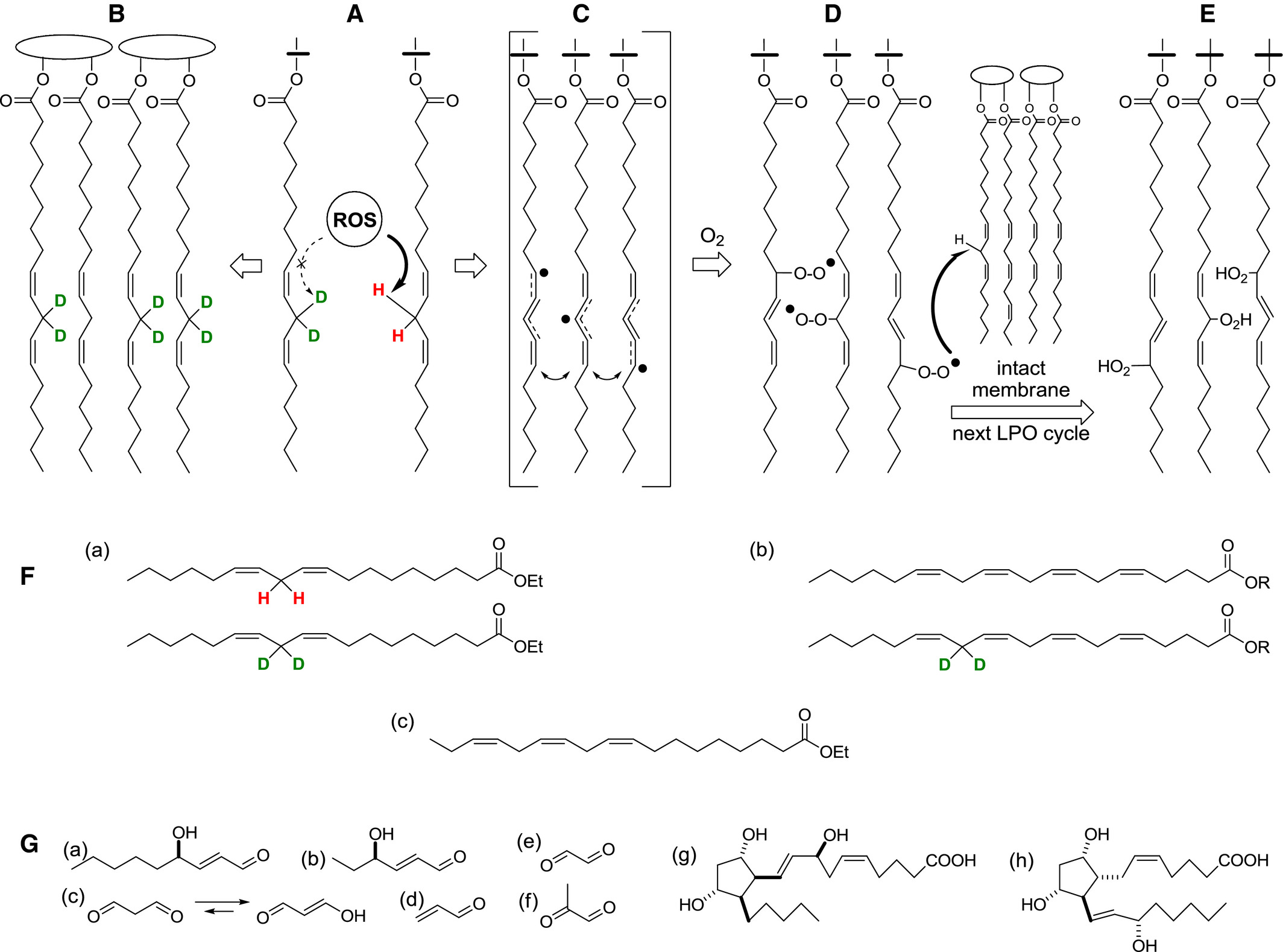
Figure 1
Protective effect of D‐PUFAs on LPO. (A) D‐PUFAs inhibit the rate‐limiting step of ROS‐driven abstraction off a bis‐allylic site. (B) A lipid bilayer that incorporates D‐PUFAs (shown, n‐6 PUFAs D2‐linoleic and D2‐ARA acids) is resistant to LPO. (C) ROS‐driven hydrogen abstraction off a bis‐allylic site (A) generates resonance‐stabilized free radicals (C), which quickly react with abundant molecular oxygen to form lipid peroxyl radicals (D). These newly formed ROS (L‐OO.) abstract H off a neighboring PUFA (turning themselves into lipid peroxides (LOOH) (E)), thus sustaining the chain reaction of LPO, which is eventually terminated by a chain‐terminating antioxidant or homologous recombination (not shown). (F), Linoleic acid or 11,11‐D2‐linoleic acid (18:2,n‐6) used in this study (a), are enzymatically converted into ARA acid and 13,13‐D2‐ARA acid (20:4,n‐6). A non‐deuterated alpha‐linolenic acid (18:3,n‐3) was used both in experimental and control diets. Once ingested, it is enzymatically converted into higher n‐3 PUFAs, EPA, and DHA (not shown). Lipid peroxides (E), which have greater volume and are more hydrophilic than nonoxidized lipids, decompose through multiple pathways (G) into numerous species such as reactive carbo‐nyls, including 4‐HNE (a), 4‐HHE (b), and MDA, which predominantly exists in one tautomeric form (c), acrylic aldehyde (d), oxalic aldehyde (e), and methylglyoxal (f). Other classes of products include ARA acid‐derived isoprostanes (g; iPF2α‐IV, or 8‐F2‐IsoP, which is one of 64 different isomers), as well as PGF2α (h), a prostaglandin that can be produced both enzymatically and nonenzymatically.
Despite the implication of oxidative stress in HD and some success in attenuating mitochondrial dysfunction in animal models, clinical trials of antioxidants for the treatment or prevention of HD have been disappointing with at best only small benefits recorded. Chain‐terminating antioxidants such as vitamin E have limited effectiveness in preventing LPO‐induced oxidative damage because of the low ratio of vitamin E in membranes relative to PUFA content (vitamin E/PUFA = 1/2000). A nonantioxidant strategy to limit LPO is to decrease the rate‐limiting initiation step of PUFA autoxidation: abstraction of bis‐allylic hydrogen atoms (Fig. 1 ). This has been accomplished by substitution of hydrogen with deuterium at bis‐allylic sites of PUFAs . This substitution (D‐PUFA versus H‐PUFA) exhibits an isotope effect on the ease of abstraction at the bis‐allylic site, resulting in a decrease in the rate of PUFA peroxidation. This strategy has been used to demonstrate partial protection of the nigrostriatal dopaminergic pathway in the MPTP mouse model of PD, to improve cholesterol handling and reduce atherosclerosis development in a hyperlipidemic mouse model of atherosclerosis, and to mitigate memory losses in the ALDH2 Alzheimer’s mouse model as well as amyloid‐beta levels in the APP/PS1 Alzheimer’s model. Both ω‐3 D4‐Lnn and ω‐6 D2‐Lin acids were employed in the above studies, while ω‐6 D2‐Lin alone was successfully used in a human Friedreich’s ataxia Phase I/II clinical study].
In the current study, we evaluated whether a diet enriched in D2‐Lin decreases LPO and provides protection against the cognitive decline present in a mouse model of HD. We have used a mouse with a human exon 1 containing approximately 140 CAG repeats ‘knocked in’ the mouse HTT gene that our laboratory has extensively characterized and used for preclinical studies. These mice have the advantage of presenting a slowly progressive phenotype, including early motor anomalies, huntingtin aggregates, and cognitive deficits, thus providing a number of endpoints with high power to detect drug effects and a suitable model for preclinical drug studies. We report that chronic administration of D2‐Lin in the diet is well‐tolerated, decreases LPO, and improves cognitive deficits in Q140 mice.
Results
One‐month‐old Q140 KI mice were administered diets containing either deuterium‐reinforced D2‐Lin (KI D‐PUFA) or control H‐Lin (KI‐cont) ethyl esters (Table 5) for 5 months. This regimen was chosen based on our previous studies showing efficient D‐PUFA incorporation by measuring deuterium content or specific D‐PUFA species on sections of the brains of mice fed a similar diet for 4–26 weeks. According to these measurements, D‐PUFA incorporation (i.e. D‐PUFA fraction of total PUFA), the 5 months long feeding would provide a level of D‐PUFA incorporation of 15–20%. This level of D‐PUFA substitution is biologically relevant, as approximately 10–20% is sufficient to decrease LPO. A cohort of wild‐type (WT) mice receiving the control diet was included to verify the presence or absence of genotypic differences.
Weight
We observed no differences in the average weights of the animals in the different treatment groups prior to euthanasia (Fig. 2). This indicates that D‐PUFA treatment did not have adverse effects on the weights of the Q140 mice.
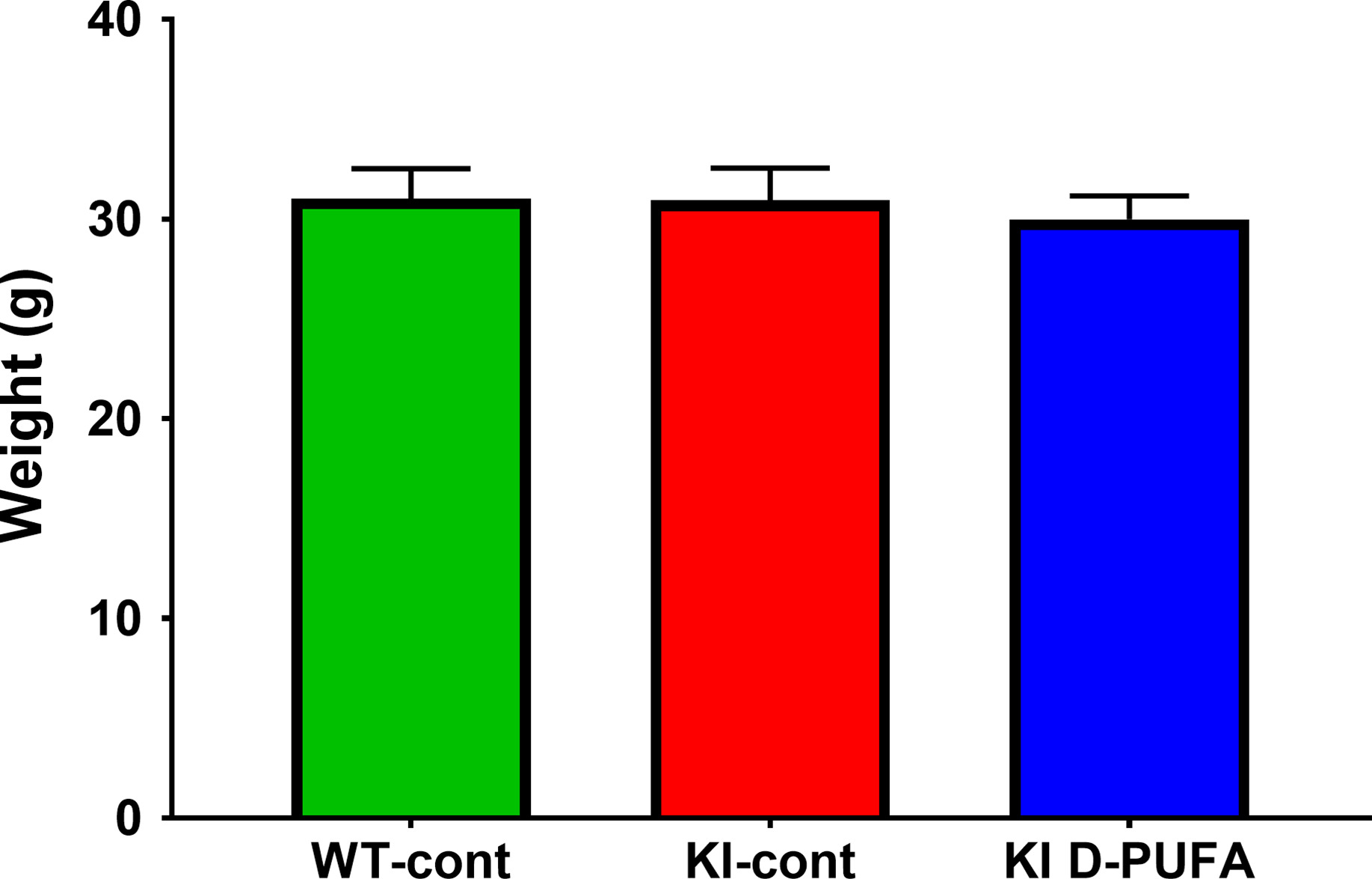
Figure 2
Weight in 6‐month‐old mice. Data are shown as the mean ± standard error of the mean. The weights of the mice in the WT‐cont (n = 17) and KI‐cont (n = 16) groups were not different (P = 0.967, Student’s t‐test). The weights of the KI mice were not modified by D‐PUFA administration (n = 18) when compared to administration of control diet (P = 0.632, Student’s t‐test).
CAG repeat length
In order to ensure that any differences between the KI‐cont and KI D‐PUFA groups were not due to differences in the number of CAG repeats, we determined the number of CAG repeats in the mice in these groups. We found no difference in the number of CAG repeats between the two groups (mean ± SEM: KI‐cont, 123.1 ± 0.7, n = 15; KI D‐PUFA, 124.1 ± 0.6, n = 18; P = 0.233 Mann–Whitney Rank Sum Test).
D‐PUFAs reduce lipid peroxidation products
As oxidative stress may be a factor in the development of HD, and since D‐PUFAs have been shown to reduce LPO products in other models, we explored whether D2‐Lin administration (Fig. 1F‐a) also reduces LPO in the Q140 KI mouse model of HD by measuring IsoP (Fig. 1G‐g) in striatal tissue as a marker for LPO. While the apparent increase in the levels of IsoP in the KI‐cont group when compared to the WT‐cont group was not significant, there was a significant decrease in IsoP levels in the KI D‐PUFA group when compared to the KI‐cont group, with D‐PUFA administration decreasing esterified F2‐isoPs by approximately 75% (Fig. 3). Thus, D‐PUFA reduced LPO in Q140 mice, confirming target engagement.
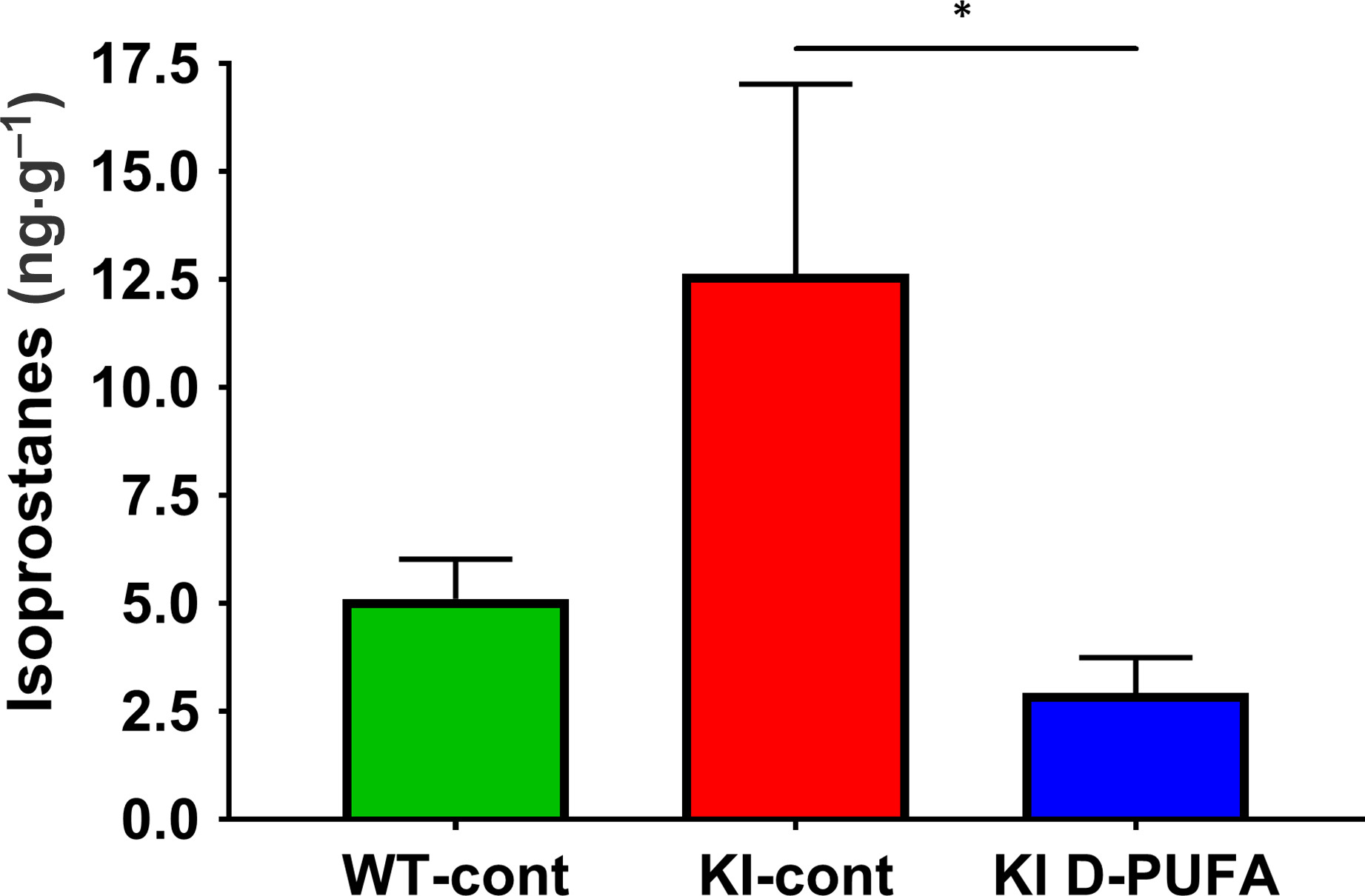
Figure 3
Isoprostane levels in striatum. Data are shown as the mean ± standard error of the mean (WT control diet: WT‐cont, n = 8; KI control diet: KI‐cont, n = 7; KI D‐PUFA n = 8). A one‐way ANOVA on Ranks of the three groups revealed a trend for a difference in isoprostanes levels between the groups (F = 5.460, P = 0.065). No genotype effect was detected by planned comparison between WT‐cont and KI‐cont (Mann–Whitney Rank Sum Test, U = 20.000, P = 0.234). However, planned comparison between KI‐cont and KI D‐PUFA revealed a decrease in isoprostanes levels in animals receiving D‐PUFA versus those receiving the control diet (Mann–Whitney Rank Sum Test U = 12.000, *P = 0.038).
We observed robust inverse correlations between IsoP levels and body weight in both KI groups, with higher IsoP levels associated with lower body weight. No such correlation was observed in the WT‐cont group (Table 1), indicating that oxidative stress in HD mice is associated with lower body weight. This negative correlation was also significant within the treated group, with those mice with the lowest IsoP levels having the highest body weights, suggesting that lowering IsoP levels by D‐PUFA may improve body weight.
Table 1. Correlation analyses between isoprostanes level, and weight and DI in the NOR
| Group | R value | P‐value | |
|---|---|---|---|
| Isoprostanes level versus weight | WT‐cont | −0.2102 | 0.651 |
| KI‐cont | −0.8432 | 0.00854 | |
| KI D‐PUFA | −0.7118 | 0.0476 | |
| Isoprostanes level versus DI | WT‐cont | −0.5075 | 0.304 |
| KI‐cont | −0.8716 | 0.00480 | |
| KI D‐PUFA | −0.7244 | 0.0656 |
- WT‐cont, wild‐type mice administered vehicle; KI‐cont, Q140 mice administered vehicle; KI D‐PUFA, Q140 mice administered deuterated polyunsaturated fatty acids. P‐values < 0.05 are indicated using bold font.
Novel object recognition
The NOR test was employed to assess the effects of D‐PUFA treatment on cognition in Q140 mice. Briefly, normal mice spend more time sniffing a novel object after prior exposure to identical objects, showing their ability to remember the familiar object by displaying a discrimination index (DI) greater than chance, chance being equal time with familiar and novel objects, represented as zero on the graph (Fig. 4). The KI mice on control diet showed a significant deficit in NOR when compared to the WT littermates on the same diet, and this deficit was reversed in mice receiving D‐PUFA (Fig. 4). Indeed, mice in the WT‐cont group but not those in the KI‐cont group displayed discrimination indices significantly higher than zero, indicating cognitive deficits in the control diet‐treated KI mice. In contrast with mice in the KI‐cont group, those in the KI D‐PUFA group performed significantly better than the chance level of zero. A strong negative correlation between DI and IsoP levels in KI‐cont mice, and a trend for a significant correlation in the KI D‐PUFA group (Table 1) suggest that cognitive impairment may be related to high oxidative stress and LPO in KI mice and that D2‐Lin may improve cognition by lowering LPO in these mice.
We observed robust inverse correlations between IsoP levels and body weight in both KI groups, with higher IsoP levels associated with lower body weight. No such correlation was observed in the WT‐cont group (Table 1), indicating that oxidative stress in HD mice is associated with lower body weight. This negative correlation was also significant within the treated group, with those mice with the lowest IsoP levels having the highest body weights, suggesting that lowering IsoP levels by D‐PUFA may improve body weight. Table 1. Correlation analyses between isoprostanes level, and weight and DI in the NOR
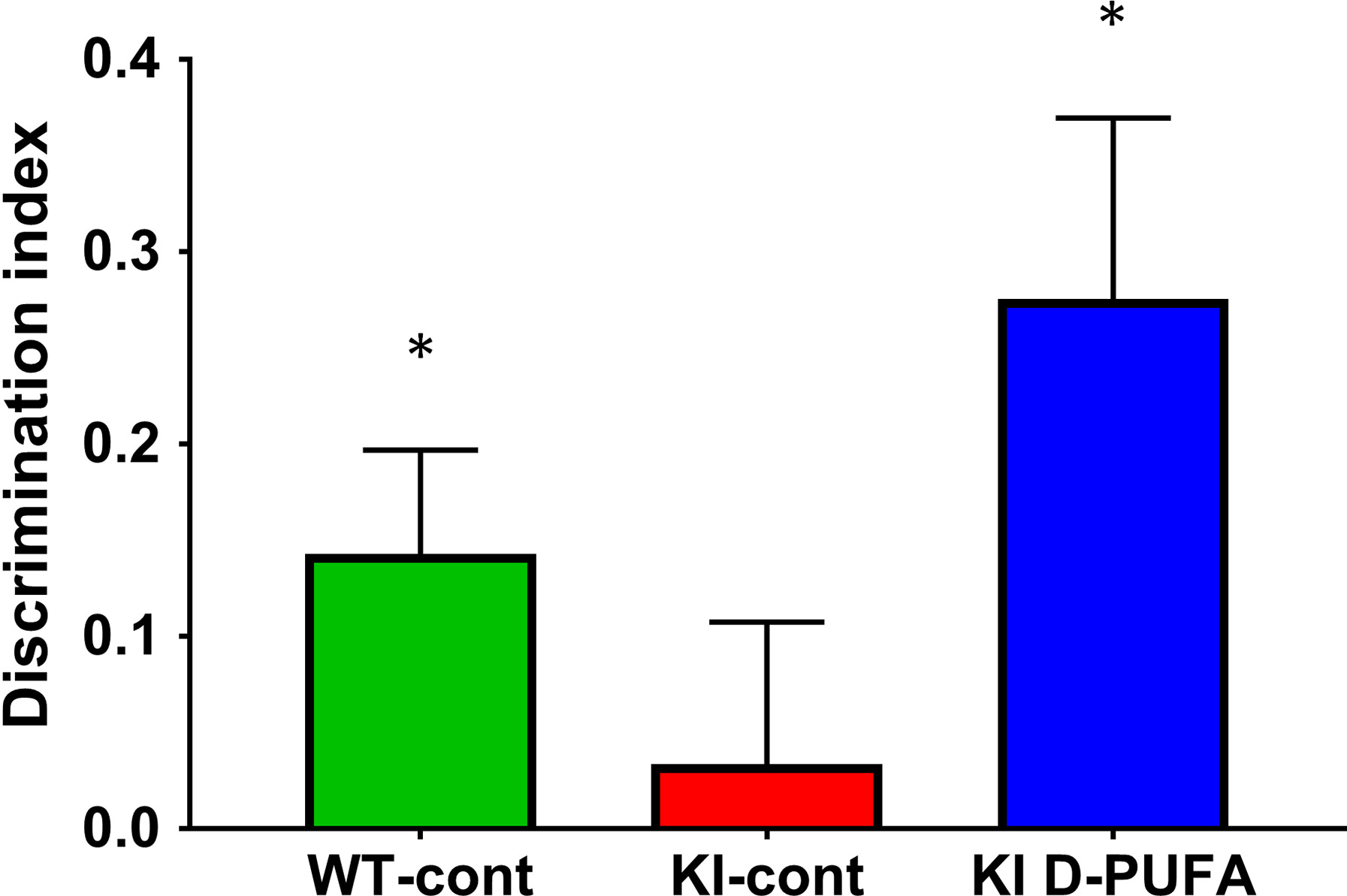
Figure 4
DI in the novel object recognition test. DI values are shown as mean ± SEM. Mice in the WT‐control diet group (WT‐cont, n = 15), but not those in the KI‐control diet group (KI‐cont, n = 16), had discrimination indices significantly higher than zero (*P = 0.0193 versus P = 0.657, respectively, one‐sample Student’s t‐test). Unlike the KI‐cont group, mice in the KI D‐PUFA group (n = 16) performed significantly better than the chance level of zero (*P = 0.0104, one‐sample Student’s t‐test).
Open field
The open field test is an automated measure of motor function. This system gathers a large number of measurements in bins of 5 min over 15 min. Only some of these measures show a genotype effect in KI‐cont mice when compared to WT mice and in general, genotype differences are most marked during the first 5 min of testing. Accordingly, we focused on measurements in this time frame to assess a possible effect of D‐PUFA. Table 2 summarizes the genotype effects observed in the different open field parameters measured in the first 5 min of the test. Genotype effects were observed, either by ANOVA and post hoc tests (move distance, distance travelled per move episode, and center entries), or by comparison with WT‐cont mice (move time and velocity). This indicates a general decrease in activity in the KI mice when compared to their WT littermates. A decrease in center entries is often considered a measure of anxiety. However, it is more likely due to the general hypoactivity of the KI mice in this study.
We observed robust inverse correlations between IsoP levels and body weight in both KI groups, with higher IsoP levels associated with lower body weight. No such correlation was observed in the WT‐cont group (Table 1), indicating that oxidative stress in HD mice is associated with lower body weight. This negative correlation was also significant within the treated group, with those mice with the lowest IsoP levels having the highest body weights, suggesting that lowering IsoP levels by D‐PUFA may improve body weight. Table 1. Correlation analyses between isoprostanes level, and weight and DI in the NOR
Figure 4
DI in the novel object recognition test. DI values are shown as mean ± SEM. Mice in the WT‐control diet group (WT‐cont, n = 15), but not those in the KI‐control diet group (KI‐cont, n = 16), had discrimination indices significantly higher than zero (*P = 0.0193 versus P = 0.657, respectively, one‐sample Student’s t‐test). Unlike the KI‐cont group, mice in the KI D‐PUFA group (n = 16) performed significantly better than the chance level of zero (*P = 0.0104, one‐sample Student’s t‐test).
Open field
The open field test is an automated measure of motor function. This system gathers a large number of measurements in bins of 5 min over 15 min. Only some of these measures show a genotype effect in KI‐cont mice when compared to WT mice and in general, genotype differences are most marked during the first 5 min of testing. Accordingly, we focused on measurements in this time frame to assess a possible effect of D‐PUFA. Table 2 summarizes the genotype effects observed in the different open field parameters measured in the first 5 min of the test. Genotype effects were observed, either by ANOVA and post hoc tests (move distance, distance travelled per move episode, and center entries), or by comparison with WT‐cont mice (move time and velocity). This indicates a general decrease in activity in the KI mice when compared to their WT littermates. A decrease in center entries is often considered a measure of anxiety. However, it is more likely due to the general hypoactivity of the KI mice in this study.
Table 2. Open field performance
| Parameter | WT‐Cont | KI‐Cont | KI D‐PUFA |
|---|---|---|---|
| Distance travelled (cm) | 983.2 ± 76.2 | *727.6 ± 60.8 | 849.6 ± 57.3 |
| Distance per move episode (cm) | 11.9 ± 1.3 | *8.2 ± 0.7 | 9.5 ± 0.7 |
| Move time (s) | 204.4 ± 7.5 | #177.6 ± 8.6 | 191.3 ± 7.1 |
| Average velocity (cm·s−1) | 3.2 ± 0.3 | #2.4 ± 0.2 | 2.8 ± 0.2 |
| Center entries | 35.706 ± 1.862 | *26.438 ± 2.347 | 31.333 ± 2.134 |
WT‐cont, wild‐type mice administered control diet; KI‐cont, Q140 mice administered control diet; KI D‐PUFA, Q140 mice administered deuterated polyunsaturated fatty acids. Distance travelled: Data are shown as the mean ± SEM (WT‐cont, n = 16; KI–cont, n = 15; KI D‐PUFA, n = 17). A one‐way ANOVA of the three groups revealed a significant difference in move distance between the groups (F = 3.722, P = 0.032). *KI‐cont had significantly shorter move distance than WT‐cont (P = 0.027, Bonferroni All Pairwise Multiple Comparisons Test). KI D‐PUFA was not different than KI‐cont (ns, Bonferroni All Pairwise Multiple Comparisons Test). Distance per move episode: Data are shown as the mean ± SEM (WT‐cont, n = 16; KI–cont, n = 15; KI D‐PUFA, n = 17). A one‐way ANOVA on Ranks of the three groups revealed a significant difference in move distance per episode between the groups (H = 6.836, P = 0.033). *KI‐cont had shorter distance per episode than the WT‐cont group (P = 0.027 Dunn’s All Pairwise Multiple Comparisons). KI D‐PUFA was not different than KI‐cont (ns, Dunn’s All Pairwise Multiple Comparisons). Move time: Data are shown as the mean ± SEM (WT‐cont n = 17; KI‐cont n = 16; KI D‐PUFA n = 18). A one‐way ANOVA of the three groups only found a nonsignificant trend for a difference in move time between the groups (F = 2.919, P = 0.064). However, planned comparisons (Student’s t‐test) confirmed a genotype effect (#P = 0.025) between the WT‐cont and KI‐cont groups but no significant effect of D‐PUFA administration (P = 0.224) compared to KI‐cont (P = 0.216). Average velocity: Data are shown as the mean ± SEM (WT‐cont, n = 16; KI‐cont, n = 15; KI D‐PUFA, n = 17). A one‐way ANOVA on Ranks of the three groups found no significant difference in average velocity between the groups (H = 1.189, P = 0.552). However, a planned comparison between on the WT‐cont and KI‐cont groups revealed a genotype effect (#P = 0.0154 Student’s t‐test). No effect of D‐PUFA administration was detected when compared to KI‐cont (P = 0.166, Student’s t‐test). Center entries: Data are shown as the mean ± SEM (WT‐cont, n = 17; KI‐cont, n = 16; KI D‐PUFA n = 18). A one‐way ANOVA of the three groups found a significant difference in center entries between the groups (F = 4.645, P = 0.014). The KI‐cont group performed less center entries than the WT‐cont group (*P = 0.011, Bonferroni All Pairwise Multiple Comparisons Test). KI D‐PUFA was not different than KI‐cont (ns, Bonferroni All Pairwise Multiple Comparisons Test).
Even though there were no longer significant differences in any of the open field measures between the mice fed D‐PUFA and WT mice fed the control diet, the means were very similar and there was no significant treatment effect when the KI D‐PUFA mice were compared to KI‐cont mice. Therefore, we conclude that D‐PUFA administration did not modify motor deficits in any significant way. Furthermore, rearing increased over 15 min in all groups (data not shown). Of note, in contrast with cognitive deficits (Table 1), move episodes, distance travelled per episode and distance travelled were not significantly correlated with isoprostanes levels in KI‐cont mice (data not shown), suggesting that, in contrast with the cognitive deficits, these motor deficits were not due to oxidative stress. It is therefore not surprising that they were not significantly improved by D‐PUFA containing diet.
Histological data
We used immunohistochemistry to assess the effects of D‐PUFA on huntingtin neuropathology in the Q140 mice. The numbers of diffuse nuclei, inclusions, nuclei with microaggregates, or neuropil aggregates per μm2 of striatal tissue did not differ between KI‐cont and KI D‐PUFA groups (Table 3).
WT‐cont, wild‐type mice administered control diet; KI‐cont, Q140 mice administered control diet; KI D‐PUFA, Q140 mice administered deuterated polyunsaturated fatty acids. Distance travelled: Data are shown as the mean ± SEM (WT‐cont, n = 16; KI–cont, n = 15; KI D‐PUFA, n = 17). A one‐way ANOVA of the three groups revealed a significant difference in move distance between the groups (F = 3.722, P = 0.032). *KI‐cont had significantly shorter move distance than WT‐cont (P = 0.027, Bonferroni All Pairwise Multiple Comparisons Test). KI D‐PUFA was not different than KI‐cont (ns, Bonferroni All Pairwise Multiple Comparisons Test). Distance per move episode: Data are shown as the mean ± SEM (WT‐cont, n = 16; KI–cont, n = 15; KI D‐PUFA, n = 17). A one‐way ANOVA on Ranks of the three groups revealed a significant difference in move distance per episode between the groups (H = 6.836, P = 0.033). *KI‐cont had shorter distance per episode than the WT‐cont group (P = 0.027 Dunn’s All Pairwise Multiple Comparisons). KI D‐PUFA was not different than KI‐cont (ns, Dunn’s All Pairwise Multiple Comparisons). Move time: Data are shown as the mean ± SEM (WT‐cont n = 17; KI‐cont n = 16; KI D‐PUFA n = 18). A one‐way ANOVA of the three groups only found a nonsignificant trend for a difference in move time between the groups (F = 2.919, P = 0.064). However, planned comparisons (Student’s t‐test) confirmed a genotype effect (#P = 0.025) between the WT‐cont and KI‐cont groups but no significant effect of D‐PUFA administration (P = 0.224) compared to KI‐cont (P = 0.216). Average velocity: Data are shown as the mean ± SEM (WT‐cont, n = 16; KI‐cont, n = 15; KI D‐PUFA, n = 17). A one‐way ANOVA on Ranks of the three groups found no significant difference in average velocity between the groups (H = 1.189, P = 0.552). However, a planned comparison between on the WT‐cont and KI‐cont groups revealed a genotype effect (#P = 0.0154 Student’s t‐test). No effect of D‐PUFA administration was detected when compared to KI‐cont (P = 0.166, Student’s t‐test). Center entries: Data are shown as the mean ± SEM (WT‐cont, n = 17; KI‐cont, n = 16; KI D‐PUFA n = 18). A one‐way ANOVA of the three groups found a significant difference in center entries between the groups (F = 4.645, P = 0.014). The KI‐cont group performed less center entries than the WT‐cont group (*P = 0.011, Bonferroni All Pairwise Multiple Comparisons Test). KI D‐PUFA was not different than KI‐cont (ns, Bonferroni All Pairwise Multiple Comparisons Test).
Table 3. Immunohistochemical analysis in KI mice
| Parameter | KI‐cont | KI D‐PUFA | KI‐cont versus KI D‐PUFA (P‐value) |
|---|---|---|---|
| Diffuse nuclei (μm−2) | 0.00706 ± 0.00055 | 0.00730 ± 0.00046 | 0.742 |
| Inclusions (μm−2) | 0.00475 ± 0.00037 | 0.00485 ± 0.00035 | 0.842 |
| Nuclei with microaggregates (μm−2) | 0.00656 ± 0.00056 | 0.00673 ± 0.00056 | 0.830 |
| Neuropil aggregates (μm−2) | 0.0138 ± 0.0021 | 0.0151 ± 0.0019 | 0.634 |
KI D‐PUFA, Q140 mice administered deuterated polyunsaturated fatty acids; KI‐cont, Q140 mice administered control diet; WT‐cont, wild‐type mice administered control diet. Data are shown as the mean ± SEM (KI‐cont, n = 9; KI D‐PUFA, n = 9). The two KI groups were compared using Student’s t‐tests. No significant differences were detected.
Discussion
Deuterium‐reinforced D‐PUFAs are resistant to nonenzymatic LPO and have been shown to reduce oxidative stress in several experimental models. In the present study, we wished to evaluate whether alteration of lipid metabolism by a D‐PUFA‐enriched diet resulted in improvement of cognitive deficits in a Q140 KI mouse model of HD. We found that oral administration of the ethyl esters of 11,11‐D2‐linoleic acid markedly reduced LPO products in Q140 KI mice, as indicated by reduced IsoP levels.
Our major finding was a significant improvement in cognitive performance in the KI‐D2‐Lin mice, as indicated by the DI in the NOR test, wherein both the WT‐cont and KI D‐PUFA groups performed significantly better than the chance level of 0, while the KI‐cont group performed at chance level. Importantly, the cognitive deficit observed in KI‐cont mice was highly correlated with the IsoP levels, supporting a role for oxidative stress in this behavioral deficit, which was improved by D‐PUFA.
D‐PUFA administration did not significantly improve motor behavior compared to the control diet, as reflected by measures in the open field test. The KI‐cont mice consistently showed decreased performance in a number of measures reflecting a general decreased activity reminiscent of the hypoactivity and akinesia that are observed in patients with HD in addition to the more typical chorea. However, these were not correlated to IsoP levels, suggesting that they may not be driven by oxidative stress and, accordingly, would not be improved by lowering LPO. The lack of significant improvement of motor deficits by D‐PUFA in our model suggests that the clinical benefits of our approach may be limited to the cognitive aspects of HD. However, it should be stressed that motor deficits in the open field may not be equivalent to the motor deficits shown by humans with HD. This is a general limitation of mouse models of motor diseases.
We designed our study to be adequately powered to detect drug effects (50% improvement at 80% power) based on our previous power calculations (Table 4), taking into account that, in order to provide both biochemical and histological postmortem data, the groups were divided for either histochemical or biochemical postmortem measurements. However, this design limited our ability to correlate behavior and levels of LPO to a subset of mice. Higher levels of IsoP were likely a reliable indicator of LPO and oxidative stress, as they were significantly associated with weight and cognitive deficits observed in KI‐cont mice, which showed a wide range of IsoP levels. In contrast, all KI mice receiving D‐PUFA had IsoP levels in the lower range, suggesting that D‐PUFA effectively decreased LPO in this mouse model of HD. Accordingly, the results of the study suggest that administration of D‐PUFA may be beneficial in HD and warrant further studies to consolidate this observation prior to testing of D‐PUFA in HD patients.
Table 4. Power analysis used to determine group sizes based on previous results
| Tests | Parameters with impairments or changes in Q140 mice (age) | n for 80% power to detect 50% improvement |
|---|---|---|
| Open field | Total move time (5 months) | 12 |
| Novel object recognition | DI (5 months) | 11 |
| Immunohistochemistry | Huntingtin aggregates (4 months) | 5 |
The present data provide useful information for the design of such additional studies. In particular, it will be important to perform experiments in older mice, as the variability of IsoP levels in KI mice fed a control diet suggest that 5–6 months of age may be just when oxidative stress develops in Q140 mice. It will be important to determine in future studies whether older mice may have more consistent levels of LPO. Furthermore, future studies should focus on biochemical postmortem measures, since we demonstrated the absence of any effect of D‐PUFA on huntingtin pathology. This is not unusual as numerous studies have found a dissociation between drug effects on huntingtin pathology and behavioral benefits and the exact role of aggregates in HD pathophysiology remains unclear.
Previous experimental studies show that antioxidant strategies protect neurons in HD models, but clinical trials with antioxidants for the treatment or prevention of HD have been consistently negative. Unfortunately, studies claiming the ineffectiveness of antioxidant interventions suffer from variations in treatment regimens and trial duration, lack of therapeutic drug monitoring, lack of monitoring oxidative stress reduction caused by drug treatment, and choice and dosage of antioxidants. For example, under some conditions, the lipophilic chain‐terminating antioxidant vitamin E can also act as a pro‐oxidant, thus serving to increase LPO. Based on data presented here and in other studies, we propose that specific inhibition of nonenzymatic LPO by D‐PUFAs may prove beneficial in a number of disease states involving LPO‐induced damage, and that this mechanism for LPO reduction is likely superior to general antioxidant inhibition or enzymatic inhibition of LPO. It is noteworthy that D‐PUFAs are not antioxidants per se: they do not quench ROS, nor do they affect cellular redox status or the antioxidant‐ROS ratio. D‐PUFAs thus allow normal ROS‐mediated signaling pathways to remain intact. It may be useful in the future, however, to determine whether combined administration of D‐PUFA with antioxidants may have additional benefits.
That D‐PUFA treatment is a viable therapeutic strategy to mitigate LPO‐induced oxidative damage in the clinical setting has very recently been evaluated in a first‐in‐human, Phase I/II clinical trial for Friedreich’s ataxia (FA), a mitochondrial disease characterized by extensive LPO. This 28‐day trial of Retrotope, Inc. (Los Altos, CA, USA) lead compound, RT001 (bideuterated linoleic acid ethyl ester), in 18 FA patients met all of its primary safety, tolerability, and pharmacodynamics goals. Given the multifactorial nature of HD, it is likely that a combination of drugs targeting different intracellular pathways in addition to mitochondrial dysfunction and oxidative stress (such as excitotoxicity, transcription dysregulation, and synaptic dysfunction) will be needed to mitigate HD symptoms. The results of the current study show that D‐PUFAs improve cognitive performance in an animal model of oxidative stress‐induced cognitive impairment by reducing LPO, and provide a rationale for future studies to determine whether D‐PUFA treatment also reduces LPO and cognitive/memory decline in humans with HD.
Materials and methods
Animal model and diet
Animal care was conducted in compliance with the United States Public Health Service Guide for the Care and Use of Laboratory Animals. The study was approved by the Institutional Animal Care and Use Committee at the University of California, Los Angeles. We used the Q140 mouse model of HD in our studies. The animals had continuous and ad libitum access to food and water. The general condition of the animals was assessed on a daily basis. Animals were housed up to five per cage in a temperature‐ and humidity‐controlled room and maintained on a 12/12 reverse light/dark cycle, with the lights on from 2200 to 1000 h. Group sizes were determined based on a power analysis of previous data from our laboratory (Table 4).
Mice were fed Research Diets’ (New Brunswick, NJ, USA) Mouse Diet AIN 93M containing defined saturated and monounsaturated fatty acids (Table 5). The PUFA component of the diet was 1.2 g (1.2%) linoleic acid and linolenic acid per 100 g diet in a 1 : 1 ratio. The experimental diet contained 11,11‐D2 linoleic acid ethyl ester (Fig. 1) and H‐α‐linolenic acid ethyl ester, whereas the control diet had the same ratios of nondeuterated, H‐PUFA. The total combined fat composition of the diet was 12.0% (saturated, monounsaturated, and PUFAs). The control and experimental diets were prepared by Research Diets with deuterated and nondeuterated D‐ and H‐PUFAs supplied by Retrotope Inc. Male and female mice were fed either D‐PUFA‐supplemented (n = 18, 10 males and eight females) or H‐PUFA‐supplemented (n = 17, eight males and nine females) diet (‘control diet’) for 5 months, beginning at 1 month of age. WT littermates were used as controls and were fed H‐PUFA‐supplemented chow (n = 18, nine males and nine females).
That D‐PUFA treatment is a viable therapeutic strategy to mitigate LPO‐induced oxidative damage in the clinical setting has very recently been evaluated in a first‐in‐human, Phase I/II clinical trial for Friedreich’s ataxia (FA), a mitochondrial disease characterized by extensive LPO. This 28‐day trial of Retrotope, Inc. (Los Altos, CA, USA) lead compound, RT001 (bideuterated linoleic acid ethyl ester), in 18 FA patients met all of its primary safety, tolerability, and pharmacodynamics goals. Given the multifactorial nature of HD, it is likely that a combination of drugs targeting different intracellular pathways in addition to mitochondrial dysfunction and oxidative stress (such as excitotoxicity, transcription dysregulation, and synaptic dysfunction) will be needed to mitigate HD symptoms. The results of the current study show that D‐PUFAs improve cognitive performance in an animal model of oxidative stress‐induced cognitive impairment by reducing LPO, and provide a rationale for future studies to determine whether D‐PUFA treatment also reduces LPO and cognitive/memory decline in humans with HD.
Table 5. Compositions of H‐ and D‐PUFA‐containing diets
| H‐diet | D‐diet | |||
|---|---|---|---|---|
| gm% | kcal% | gm% | kcal% | |
| D‐linoleic acid, ethyl ester | – | – | 0.6 | 1.2 |
| H‐linoleic acid, ethyl ester | 0.6 | 1.2 | – | – |
| H‐linolenic acid, ethyl ester | 0.6 | 1.2 | 0.6 | 1.2 |
| Oleate, ethyl | 2.9 | 6.2 | 2.9 | 6.2 |
| Coconut oil, 101 (hydrogenated) | 7.5 | 16.0 | 7.5 | 16.0 |
| Casein | 13.5 | 12.8 | 13.5 | 12.8 |
| L‐cystine | 0.0 | 0.2 | 0.0 | 0.2 |
| Corn starch | 43.2 | 40.9 | 43.2 | 40.9 |
| Maltodextrin 10 | 12.1 | 11.4 | 12.1 | 11.4 |
| Sucrose | 9.7 | 9.1 | 9.7 | 9.1 |
| Cellulose | 4.8 | 0.0 | 4.8 | 0.0 |
| t‐Butylhydroquinone | 0.0 | 0.0 | 0.0 | 0.0 |
| Mineral Mix S10022M | 3.4 | 0.0 | 3.4 | 0.0 |
| Vitamin Mix V10037 | 1.0 | 0.9 | 1.0 | 0.9 |
| Choline bitartrate | 0.2 | 0.0 | 0.2 | 0.0 |
a AIN‐93M rodent‐based diets were modified to contain 12% fat with 1.2% D‐linoleic : H‐linolenic (1 : 1 weight ratio), or 1.2% H‐linoleic : H‐linolenic (1 : 1 weight ratio). Bold font is used to indicate differences between the two diets used in the study.
Behavioral analyses
All testing was carried out blinded to genotype and treatment. The mice were tested in the light phase, and were first habituated to the procedure room for 15 min prior to testing. All behavior tests were performed at the age of 6 months following 5 months of treatment.
Open field
We used an automated activity monitoring apparatus to assess the motor behavior of the mice 1 week before the end of the study (Truscan, Coulbourn Instruments, Allentown, PA, USA). The mice were placed individually into the apparatus and different parameters of their activity were monitored for 15 min (divided into three 5‐min bins) using infra‐red beams. The apparatus was cleaned using 70% ethanol between testing individual mice.
Novel object recognition
The open field apparatus was used for this test. The NOR test was performed 24 h after the open field test and was carried out over 2 days. A video camera was used to record the mice for 10 min on each day. On the first day, two identical objects (plastic geometrical forms) were placed in the open field arena. The mouse was then placed in the arena and allowed to investigate the objects for 10 min. On the second day, one of the previously explored objects (familiar object: F) was replaced with a novel object (N) with a different shape and color. The mouse was again placed in the arena and recorded for 10 min. At the conclusion of the test, the video recordings were reviewed and the time spent sniffing each object during the first 5 min of the test was recorded by an investigator blind to genotype and treatment. The apparatus and objects were cleaned between trials using 70% ethanol. The DI was calculated as (N−F)/(N+F). As such, a positive DI reflected a preference for the novel object, while a DI of zero reflected chance performance in the test.
Biochemical measurements
At the end of the study, half the mice from each group were anesthetised with pentobarbital and trans‐cardially perfused with PBS. The brain was removed, dissected, and frozen in liquid nitrogen until analysis.
The LPO markers (esterified) F2‐IsoPs and prostaglandin F2α (PGF2α) were determined in cortex and hippocampus at the Eicosanoid Core Laboratory at Vanderbilt University Medical Center. Samples were pretreated and analysed for (esterified) F2‐IsoPs and PGF2α using GC/negative ion chemical ionization MS as described in detail previously .
Histology
At 6 months of age, half of the mice from each group were anesthetized and perfused with 4% paraformaldehyde (PFA), their brains removed, post fixed for 8 h in 4% PFA, cryoprotected in 30% sucrose and frozen for later use. Brain sections (40 μm thick) were immunostained with the monoclonal antibody EM48 to detect nuclear staining and Htt aggregates (modified from Ref.). The sections were washed in 0.01 m PBS and endogenous peroxidases were inactivated by incubating the sections in 1% H2O2 and 0.5% Triton X‐100 in PBS for 20 min. Nonspecific binding was blocked by incubating the sections for 30 min at room temperature in PBS containing 3% bovine serum albumin (BSA) and 2% normal goat serum (NGS). The primary antibody EM48 was diluted (1 : 300) in PBS containing 3% BSA, 2% NGS, 0.08% sodium azide, and 0.2% Triton X‐100. The sections were incubated in the primary antibody solution overnight at room temperature. The sections were then washed in PBS and incubated in biotinylated goat anti‐mouse antibody (1 : 200; Vector ABC Elite; Vector, Burlingame, CA, USA) for 1 h at room temperature. The sections were washed again and incubated in avidin–biotin complex (Vector ABC Elite) in PBS containing 0.2% Triton X‐100 for 2 h. Immunoreactivity was visualized by incubation in 0.03% 3‐3‐diaminobenzidine tetrahydrochloride (Sigma, St. Louis, MO, USA) and 0.0006% H2O2 in 0.05 m Tris buffer, pH 7.6. The sections were mounted on slides after five washes in Tris buffer. Control sections were processed in parallel in the absence of the primary or secondary antibodies.
In order to quantify the huntingtin aggregates, the numbers of stained nuclei containing microaggregates, neuropil aggregates, inclusions, and diffuse nuclei were counted using stereoinvestigator software (Microbrightfield, Colchester, VT, USA), as previously described in Refs . All analyses were performed by an investigator unaware of genotype and treatment.
Data analysis
Data are expressed as the mean ± standard error of the mean and were analyzed using one‐way ANOVAs, one‐way ANOVA on Ranks, one‐way repeated measures ANOVAs, Student’s t‐tests, or Mann–Whitney Rank Sum tests, as indicated. Nonparametric tests (Mann–Whitney Rank Sum test or ANOVA on Ranks) were used when non‐normal data (P < 0.050 in Shapiro–Wilk test) were present. The study was designed to evaluate both genotype (WT versus KI, both on control diet) and treatment effects (KI‐cont versus KI‐D‐PUFA groups) and was powered based on our previous data for two‐group comparisons. Therefore, we performed planned comparisons between these groups to assess genotype and treatment effects, as indicated. ANOVAs were performed as additional exploratory analyses to compare all three groups. Outliers were excluded based on the results of Grubbs’ tests. A P‐value of 0.05 or less was considered statistically significant. All statistical analyses were performed using sigmaplot (Systat Software, Inc., San Jose, CA, USA).
Acknowledgments
This study was funded by a grant from the Hereditary Disease Foundation and the Charles H. Markham endowed chair to MFC. The authors gratefully acknowledge the support of Dr M. Levine, the Hereditary Disease Foundation, CHDI, Inc. and PHS grant NS41574 for the Q140 colony at UCLA. Retrotope Inc. provided D‐ and H‐PUFAs for this study.
Conflict of interest
CRC and MSS are shareholders of Retrotope, Inc.
Author contributions
AH managed the study, performed and supervised the experiments, analyzed the results, performed statistical analyses, and prepared the figures and the manuscript; CZ and ARG performed experiments, supervised data gathering, analyzed data, and contributed portions of the manuscript; CE, AG, MJ, and GM performed experiments and data analyses, and reviewed the manuscript; MFC designed the experiments, supervised execution, analyzed results, and edited the manuscript; MSS designed the experiments, analyzed results, contributed portions of, and edited the manuscript.